Egyes gépitanulás-modellek ugyanúgy szekvenciába tudják kódolni az aminosavak „nyelvtanát”, mint ahogy a nyelvtannal mondatokká válnak a szavak. A Salesforce Research és az Illinois Egyetem (Urbana-Champaign) kutatói ilyen – fontos biológiai jellemzőket kimutató – modellek interpretálására fejlesztettek módszereket.
Mélytanulással működő modelleket eddig is gyakoroltattak a fehérjéket alkotó aminosav-szekvenciákon, és sikeresek voltak: működő szekvenciákat csoportosítottak, generáltak. Az új kutatás eggyel előrébb lép, mert a modellek a fehérjeszerkezetről is szolgáltatnak információkat.
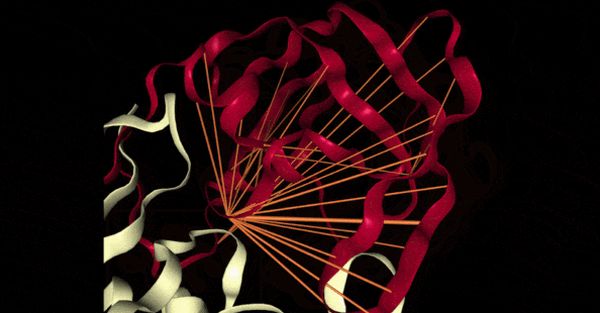
Amikor aminosavak összekapcsolódnak egymással, a szekvencia formája meghatározza a kialakuló fehérje biológiai funkcióját. Ezeken a szekvenciákon gyakoroltatott gépitanulás-modellek a két sav közötti bizonyos értékek alapján meg tudják határozni, hogy fontos szerepet játszanak a fehérje szerkezetében, például kapcsolatba lép más fehérjékkel.
A kutatók aminosav-szekvenciák adatbázisán treníroztatták modelljüket, hogy aztán előrejelezze a működésüket. Egy megadott szint alatti értékeket eleve kiszűrtek ahhoz, hogy az erős kapcsolatban álló savpárokat tudják tanulmányozni. Az adatbázisban lévő információk alapján összeszámolták a fehérje formájához társított kapcsolatokat, például az érintkező savpárokat.
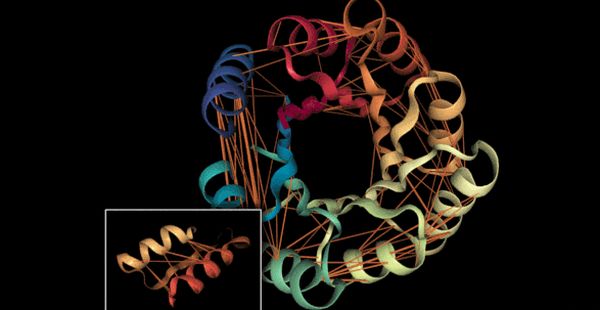
Egyes tulajdonságok csak a pár egyik felétől függtek.
Eredményeiket összehasonlították más fehérje-adatbázisokkal, az azokból levont következtetésekkel. Kiderült, hogy modelljük alsó rétegeiben jelentős mértékben nőtt a tényleges kapcsolatban álló, rokon aminosav-párok száma (44,7 százalék), míg az összes aminosav esetében ez a mutató csak 1,3 százalék volt.
Annak az esélye, hogy egy rokon pár második aminosavától is függnek fehérjék tulajdonságai, rétegről rétegre nem emelkedett jelentősen, de így is 48,2 százalékot ért el. Összehasonlításként: bármely aminosav esetében ez mindössze 4,8 százalék.
Egy szekvenciából hiányzó aminosavak előrejelzésével fontos dolgokat tudunk meg arról, hogy a savak hogyan állnak össze nagyobb szerkezetekké, és így nemcsak a modell, hanem a természet működéséről is bővülnek az ismereteink.
Ilyen modellekkel vírusok, például a SARS-CoV-2 szerkezetéről is több információt gyűjthetünk össze.